4.2 Analyzing viral protein sequences
Let’s practice with starting a project.
Viral structural proteins are dramatically more important to the general public than they were 2 years ago - and under much more public attention. Let’s take a cursory look at some of these.
What kind of data are we using for this? We’ll retrieve some determined protein sequences from NCBI databases. We’ve already done that work for you already. Here’s a short writeup of what we did, amd we would recommend that you always document where the data came from and how it was retrieved/processed:
The NCBI Identical Protein Groups database was queried for “structural” with “Division” restricted to “Viruses” and “Source database” restricted to “UniProtKB/Swiss-Prot”. 243 results were retrieved as FASTA files and converted using bash commands into separate tab-delimited files. 3
What format are these in?
These files are in the data/viral_structural_proteins folder,
they end in .tsv, are tab-delimited three fields, and look like this:
Q8V433.1 Membrane protein Bovine respiratory coronavirus (strain 98TXSF-110-LUN) MSSVTTPAPVYTWTA...This is not a standard format, because I’d like to tell y’all about reading tab-delimited files. 4
One trick to look at files using R is using readLines() function.
This just reads lines, up to the argument n= number of lines.
Try something like:
## [1] "YP_009724393.1 membrane glycoprotein \tSevere acute respiratory syndrome coronavirus 2\tMADSNGTITVEELKKLLEQWNLVIGFLFLTWICLLQFAYANRNRFLYIIKLIFLWLLWPVTLACFVLAAVYRINWITGGIAIAMACLVGLMWLSYFIASFRLFARTRSMWSFNPETNILLNVPLHGTILTRPLLESELVIGAVILRGHLRIAGHHLGRCDIKDLPKEITVATSRTLSYYKLGASQRVAGDSGFAAYSRYRIGNYKLNTDHSSSSDNIALLVQ"Whenever using these commands, put your cursor in the quotes and then hit TAB, to use autocomplete.
Let’s do two things. Let’s count how long the protein sequences are, so we can look at that distribution. Let’s also look for particular combinations of amino acids.
Some folks are quite interested in the insertion of an amino-acid sequence
of PRRA, relative to other related coronaviruses, in the SARS-CoV-2 genome.
I don’t know hardly anything about furin-cleavage sites or viral evolution,
but since this is an introductory R class,
we can use the tools we have to look for this sequence in the files.
Let’s do that5
How?
4.2.1 Reading in tab-delimited file
Let’s read one of them into R.
The records are “delimited” by tabs, so each field is tab-separated.
We’ll need to use the read.delim function and specify a tab separator.
4.2.1.1 Review of looking up documentation
You can look up the documentation for any named function or package by using
the ?function syntax.
Sometimes, you need to use extra backticks to make it work, like ?`+`
If you don’t know what you’re looking for, you can search with ??.
Extra backticks don’t hurt, and are necessary when you have spaces in the
query.
Once the documentation is open, you can search for text.
If you are using the R console, you can use / to open a search bar and
enter-key to search for it.
4.2.1.2 Back to the file-reading…
We can use read.delim to read in one of the files.
You will have to specify the delimiter/separator.
" is usually how you specify a TAB character in character strings, like
you would set as that argument.
viral_protein_data <- read.delim("data/viral_structural_proteins/viral_proteins_100.tsv",
sep="\t",header=F)
viral_protein_data## V1
## 1 Q8V433.1 Membrane protein
## V2
## 1 Bovine respiratory coronavirus (strain 98TXSF-110-LUN)
## V3
## 1 MSSVTTPAPVYTWTADEAIKFLKEWNFSLGIILLFITVILQFGYTSRSMFVYVIKMIILWLMWPLTIILTIFNCVYALNNVYLGFSIVFTIVAIIMWIVYFVNSIRLFIRTGSWWSFNPETNNLMCIDMKGRMYVRPIIEDYHTLTVTIIRGHLYMQGIKLGTGYSLSDLPAYVTVAKVSHLLTYKRGFLDKIGDTSGFAVYVKSKVGNYRLPSTQKGSGLDTALLRNNIWhat type is that third column?
## [1] "factor" "integer" "oldClass"
## [4] "double" "numeric" "vector"
## [7] "data.frameRowLabels"Huh, looks like it’ll convert it to a factor automatically. Let’s tell it to
not do that, to leave it as.is.
viral_protein_data <- read.delim("data/viral_structural_proteins/viral_proteins_100.tsv",
sep="\t",header=F,as.is=T)
viral_protein_data## V1
## 1 Q8V433.1 Membrane protein
## V2
## 1 Bovine respiratory coronavirus (strain 98TXSF-110-LUN)
## V3
## 1 MSSVTTPAPVYTWTADEAIKFLKEWNFSLGIILLFITVILQFGYTSRSMFVYVIKMIILWLMWPLTIILTIFNCVYALNNVYLGFSIVFTIVAIIMWIVYFVNSIRLFIRTGSWWSFNPETNNLMCIDMKGRMYVRPIIEDYHTLTVTIIRGHLYMQGIKLGTGYSLSDLPAYVTVAKVSHLLTYKRGFLDKIGDTSGFAVYVKSKVGNYRLPSTQKGSGLDTALLRNNI## [1] "V1" "V2" "V3"## [1] "character" "vector" "data.frameRowLabels"
## [4] "SuperClassMethod"Note that you can find the column names, or names of columns, using colnames.
Or names. And you can inspect the type in several different ways.
This is how you do it in base R. How do you do this in tidyverse?
You can use read_delim or read_tsv, with different arguments,
such as col_names=F to prevent it from reading the first line as a header.
Also, no factor conversion :)
4.2.2 String stuff
Once we have that, we can test how we will process the protein sequence. For now, let’s just calculate the length of the protein sequence.
Character strings are a common type of data, and there’s lots of ways to cut, dice, extract, and recognize elements to help your work. Here’s some tools/ideas:
4.2.2.1 Base R tools
Let’s start with one of those above sentences. You can make a string by putting characters in between single or double quotes:
## [1] "Character strings are a common type of data, and there's lots of ways to"## [1] "character" "vector" "data.frameRowLabels"
## [4] "SuperClassMethod"## chr "Character strings are a common type of data, and there's lots of ways to"grep and grepl are good for searching for patterns (like grep in bash).
The first returns position, the second returns TRUE or FALSE.
I only ever use grepl anymore. It searches for a pattern string
in a character vector x, and returns a logical vector.
With a logical vector, you can do a lot, like indexing/slicing or sum-ing.
## [1] TRUE## [1] FALSE## [1] TRUESo see that last one with the []* stuff?
Called a regular expression.
You can use complex
regular expressions
to specify more complex patterns.
These can get
really complex
and really powerful.
We’re not going to explain these here, except just for the example below.
Ask on Slack later if you’d like more help with something specific.
Also of use is sub and gsub.
These substitute patterns with replacements, for the
first occurrence (sub) or globally (gsub).
These are really handy for modifying data-tables.
For example, let’s say you’d like to split up filenames by dates:
datar <- data.frame(stringsAsFactors=F,
filenames=c(
"210607_pilot_transformation_works.jpeg",
"210609_transformation_seems_to_work.jpeg",
"210610_failed_gel_images.jpeg"
)
)
datar$dates <- gsub(pattern="(\\d\\d\\d\\d\\d\\d)_(.*)\\.jpeg",
replacement="\\1",x=datar$filenames)
datar$names <- gsub(pattern="(\\d\\d\\d\\d\\d\\d)_(.*)\\.jpeg",
replacement="\\2",x=datar$filenames)
datar## filenames dates names
## 1 210607_pilot_transformation_works.jpeg 210607 pilot_transformation_works
## 2 210609_transformation_seems_to_work.jpeg 210609 transformation_seems_to_work
## 3 210610_failed_gel_images.jpeg 210610 failed_gel_imagesThe above uses regular expressions, where \\d is matching any digit,
.* matches anything, and () denote “groups” to “capture”.
In the “replacement”, these “groups” are referred to by \\1 or \\2.
These “capture groups” are super useful! You can use them to take out matches and re-arrange them.
So, (\\d\\d\\d\\d\\d\\d)_ matches six digits followed by an underscore,
such as 210618_, and saves it as \\1. So
## [1] "210616"## [1] "after"There are better (cleaner) ways of doing this with tidyverse, basics are important. See a bit of tidyverse/stringr stuff in the end of this 4.2 section, if you’re curious.
You should now be able to manipulate within character strings a bit, identify and replace patterns.
4.2.3 Loops - doing a similar task multiple times
You will want to repeat this analysis for all the files. The simplest way of doing this is to copy and paste it, and change the filename.
viral_protein_data <- read.delim("data/viral_structural_proteins/viral_proteins_002.tsv",
sep="\t",header=F, as.is=T)
nchar(viral_protein_data$V3)## [1] 177viral_protein_data <- read.delim("data/viral_structural_proteins/viral_proteins_003.tsv",
sep="\t",header=F, as.is=T)
nchar(viral_protein_data[1,"V3"])## [1] 137Go ahead and do this for all 242 proteins
… just kidding.
This is a lot of work, and each time we do this we can introduce errors. If we ever have more files, we have to copy and paste more. If we ever want to change an argument for all of them, we have to do each one.
Instead, we can work with a list of all the files available:
## [1] "viral_proteins_000.tsv" "viral_proteins_001.tsv" "viral_proteins_002.tsv"
## [4] "viral_proteins_003.tsv" "viral_proteins_004.tsv"Note that I put a [1:5] to limit it to the first 5.
You could also use head(). It’s a good idea to work
with a small subset of files while you are iterating
through development, then scale it up to the entirety.
But watch out…
## [1] "viral_proteins_238.tsv" "viral_proteins_239.tsv"
## [3] "viral_proteins_240.tsv" "viral_proteins_241.tsv"
## [5] "viral_proteins_242.tsv" "viral_structural_proteins.fasta"At the very end is another different file, a FASTA file.
So let’s use a pattern
argument in list.files to specify what we want to list.
And we’ll get back to the FASTA later…
## [1] "viral_proteins_237.tsv" "viral_proteins_238.tsv" "viral_proteins_239.tsv"
## [4] "viral_proteins_240.tsv" "viral_proteins_241.tsv" "viral_proteins_242.tsv".* stands for 0 or more (*) of anything (.). For more details,
look up regular-expressions.
4.2.3.1 What are loops?
Loops are for running a “code block” as many times as the “condition” determines.
A “code block” is either one line of code, or multiple lines of code surrounded by curly brackets - {}
{ code <- "in a block" with <- "multiple lines" }The code just runs. Yep. It’s that simple.
A “condition” is an expression of code that can either evaluate to either
TRUEorFALSE, or set a variable for each time running the code block. This is often just before the code block, in()parentheses.
The most common form of these kind of “control statements” is a for loop.
Other “control statements” or “flow control statements” are
while, repeat, and if.
Let’s look up what they do, with ?`for`
Note the back ticks! These are a trick in R to make anything
be interpreted as literally what you type, and not
any special characters. Like ?`+`, or ?`?`
Here’s an example loop:
## [1] 1
## [1] 2
## [1] 3
## [1] 4The pieces:
(i in 1:4)is what is being looped over -1:4is a vector of 1 through 4 that is created, and it is put one at a time intoi(a new variable). You need the parentheses.{and}denote the opening and closing brackets, specify the “code block” that is run each time.inside this “code block” is
print(i)- it prints the variablei, which is set to a value of 1, 2, 3, or 4 for each loop
## [1] "viral_proteins_000.tsv"
## [1] "viral_proteins_001.tsv"
## [1] "viral_proteins_002.tsv"
## [1] "viral_proteins_003.tsv"
## [1] "viral_proteins_004.tsv"Inside the code block we can do more than print, like looking for a motif. Here, we’ll try to read the file and take a look at the sequence …
for (i in list.files(path="data/viral_structural_proteins",
pattern=".*tsv")[1:5] ) {
print(i)
print(read.delim(i)$V3)
}## [1] "viral_proteins_000.tsv"## Warning in file(file, "rt"): cannot open file 'viral_proteins_000.tsv': No such
## file or directory## Error in file(file, "rt"): cannot open the connectionError! It is looking for a file viral_proteins_000.tsv, but it is looking
in this directory. It is actually in data/viral_structural_proteins.
Look up the list.files documentation, and find how to get it to return
the full name of the file.
Next, this should work…
for (i in list.files(path="data/viral_structural_proteins",
pattern=".*tsv",full.names=T)[1:5] ) {
print(read.delim(i,sep="\t",header=F,as.is=T)$V3)
}## [1] "MEFIPTQTFYNRRYQPRPWTPRPTIQVIRPRPRPQRQAGQLAQLISAVNKLTMRAVPQQKPRRNRKNKKQKQKQQAPQNNTNQKKQPPKKKPAQKKKKPGRRERMCMKIENDCIFEVKHEGKVTGYACLVGDKVMKPAHVKGTIDNADLAKLAFKRSSKYDLECAQIPVHMKSDASKFTHEKPEGYYNWHHGAVQYSGGRFTIPTGAGKPGDSGRPIFDNKGRVVAIVLGGANEGARTALSVVTWNKDIVTKITPEGAEEWSLAIPVMCLLANTTFPCSQPPCIPCCYEKEPEETLRMLEDNVMRPGYYQLLQASLTCSPHRQRRSTKDNFNVYKATRPYLAHCPDCGEGHSCHSPVALERIRNEATDGTLKIQVSLQIGIGTDDSHDWTKLRYMDNHIPADAGRAGLFVRTSAPCTITGTMGHFILARCPKGETLTVGFTDSRKISHSCTHPFHHDPPVIGREKFHSRPQHGKELPCSTYVQSNAATAEEIEVHMPPDTPDRTLLSQQSGNVKITVNGRTVRYKCNCGGSNEGLITTDKVINNCKVDQCHAAVTNHKKWQYNSPLVPRNAELGDRKGKIHIPFPLANVTCMVPKARNPTVTYGKNQVIMLLYPDHPTLLSYRSMGEEPNYQEEWVTHKKEVVLTVPTEGLEVTWGNNEPYKYWPQLSANGTAHGHPHEIILYYYELYPTMTVVVVSVASFILLSMVGMAVGMCMCARRRCITPYELTPGATVPFLLSLICCIRTAKAATYQEAAVYLWNEQQPLFWLQALIPLAALIVLCNCLRLLPCCCKTLAFLAVMSIGAHTVSAYEHVTVIPNTVGVPYKTLVNRPGYSPMVLEMELLSVTLEPTLSLDYITCEYKTVIPSPYVKCCGTAECKDKNLPDYSCKVFTGVYPFMWGGAYCFCDAENTQLSEAHVEKSESCKTEFASAYRAHTASASAKLRVLYQGNNITVTAYANGDHAVTVKDAKFIVGPMSSAWTPFDNKIVVYKGDVYNMDYPPFGAGRPGQFGDIQSRTPESKDVYANTQLVLQRPAAGTVHVPYSQAPSGFKYWLKERGASLQHTAPFGCQIATNPVRAMNCAVGNMPISIDIPDAAFTRVVDAPSLTDMSCEVPACTHSSDFGGVAIIKYAVSKKGKCAVHSMTNAVTIREAEIEVEGNSQLQISFSTALASAEFRVQVCSTQVHCAAECHPPKDHIVNYPASHTTLGVQDISATAMSWVQKITGGVGLVVAVAALILIVVLCVSFSRH"
## [1] "MFPFQPMYPMQPMPYRNPFAAPRRPWFPRTDPFLAMQVQELTRSMANLTFKQRRGAPPEGPPAKKSKREAPQKQRGGQRKKKKNEGKKKAKTGPPNLKTQNGNKKKTNKKPGKRQRMVMKLESDKTFPIMLEGKINGYACVVGGKLFRPMHVEGKIDNDVLAALKTKKASKYDLEYADVPQNMRADTFKYTHEKPQGYYSWHHGAVQYENGRFTVPRGVGARGDSGRPILDNQGRVVAIVLGGVNEGSRTALSVVMWNEKGVTVKYTPENCEQWSLVTTMCLLANVTFPCAQPPICYDRKPAETLAMLSANVDNPGYDELLKAAVTCPGRKRRSTEELFKEYKLTRPYMARCVRCAVGSCHSPIAIEAVKSDGHDGYVRLQTSSQYGLDPSGNLKSRTMRYNMYGTIEEIPLHQVSLHTSRPCHIVDGHGYFLLARCPAGDSITMEFKKDSVTHSCSVPYEVKFNPVGRELYTHPPEHGAEQACQVYAHDAQNRGAYVEMHLPGSEVDSSLVSLSSGLVSVTPPAGTSALVECECSGTTISKTINKTKQFSQCTKKEQCRAYRLQNDKWVYNSDKLPKAAGATLKGKLHVPFLLADGKCTVPLAPEPMITFGFRSVSLKLHPKYPTYLTTRELADEPHYTHELISEPSVRNFSVTAKGWEFVWGNHPPKRFWAQETAPGNPHGLPHEVIVHYYHRYPMSTITGLSICAAIVAVSIAASTWLLCRSRASCLTPYRLTPNAKMPLCLAVLCCARSARAETTWESLDHLWNNNQQMFWTQLLIPLAALIVVTRLLKCMCCVVPFLVVAGAAGAGAYEHATTMPNQAGISYNTIVNRAGYAPLPISITPTKIKLIPTVNLEYVTCHYKTGMDSPTIKCCGSQECTPTYRPDEQCKVFAGVYPFMWGGAYCFCDTENTQISKAYVMKSEDCLADHAAAYKAHTASVQALLNITVGEHSTVTTVYVNGETPVNFNGVKLTAGPLSTAWTPFDRKIVQYAGEIYNYDFPEYGAGQPGAFGDIQLRTVSSSDLYANTNLVLQRPKAGAIHVPYTQAPSGFEQWKKDKAPSLKFTAPFGCEIYTNPIRAENCAVGSIPLAFDIPDALFTRVSETPTLSAAECTLNECVYSSDFGGIATVKYSASKSGKCAVHVPSGTATLKEASVELAEQGSVTIHFSTANIHPEFRLQICTSFVTCKGDCHPPKDHIVTHPQYHAQTFTAAVSKTAWTWLTSLLGGSAVIIIIGLVLATLVAMYVLTNQKHN"
## [1] "MFNIKMTISTLLIALIILVIIILVVFLYYKKQQPPKKVCKVDKDCGSGEHCVRGTCSTLSCLDAVKMDKRNIKIDSKISSCEFTPNFYRFTDTAADEQQEFGKTRHPIKITPSPSESHSPQEVCEKYCSWGTDDCTGWEYVGDEKEGTCYVYNNPHHPVLKYGKDHIIALPRNHKHA"
## [1] "MEAVLTKLDQEEKKALQNFHRCAWEETKNIINDFLEIPEERCTYKFNSYTKKMELLFTPEFHTAWHEVPECREFILNFLRLISGHRVVLKGPTFVFTKETKNLGIPSTINVDFQANIENMDDLQKGNLIGKMNIKEG"
## [1] "MAFLMSEFIGLGLAGAGVLSNALLRRQELQLQKQALENGLVLKADQLGRLGFNPNEVKNVIVGNSFSSNVRLSNMHNDASVVNAYNVYNPASNGIRKKIKSLNNSVKIYNTTGESSV"And yep, we have protein sequences. Now looking for some amino acids:
for (i in list.files(path="data/viral_structural_proteins",
pattern=".*tsv",full.names=T)[1:5] ) {
print(grepl("QQ",read.delim(i,sep="\t",header=F,as.is=T)$V3))
}## [1] TRUE
## [1] TRUE
## [1] TRUE
## [1] FALSE
## [1] FALSE4.2.4 Storing values from a loop
First, how can we store the filenames and use them later for the loop? We need to turn out list of files into indicies. We’ll save it first so we can count how many there are.
seq_along is handy function to create a number sequence along a vector,
otherwise use something like seq(1,length(x)).
# listing files
first_five <- list.files(path="data/viral_structural_proteins",pattern=".*tsv")[1:5]
length(first_five)## [1] 5## [1] 1
## [1] "viral_proteins_000.tsv"
## [1] 2
## [1] "viral_proteins_001.tsv"
## [1] 3
## [1] "viral_proteins_002.tsv"
## [1] 4
## [1] "viral_proteins_003.tsv"
## [1] 5
## [1] "viral_proteins_004.tsv"Okay, but how do we store values for later analysis?
In other languages, “append”. But R is not built that way. It’ll work
(using something like append, or x <- c(x, new_value)),
but it’s inefficient. The “R-way” to store values from a loop
is to define a vector of the right length, then put each element in it.
Here’s some example vectors that we can create.
## [1] "" "" "" "" "" "" "" "" "" ""## [1] 0 0 0 0 0## [1] FALSE FALSEYou can save these to a variable, and thus have a character vector, numeric vector, or logical vector, of different sizes.
Putting these together, we can create and save a vector of file names:
# listing files
first_five <- list.files(path="data/viral_structural_proteins",pattern=".*tsv")[1:5]
# initializing vectors
filenamez <- vector("character",length(first_five))
# looping through files
for (i in seq_along(first_five) ) {
filenamez[i] <- first_five[i]
}
filenamez## [1] "viral_proteins_000.tsv" "viral_proteins_001.tsv" "viral_proteins_002.tsv"
## [4] "viral_proteins_003.tsv" "viral_proteins_004.tsv"And finally calculate the length of each protein: And
# listing files
first_five <- list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")[1:5]
# initializing vectors
has_qq <- vector("logical",length(first_five))
# looping through files
for (i in seq_along(first_five) ) {
# calculating length and storing it
has_qq[i] <- grepl( "QQ", read.delim(first_five[i],
header=F,as.is=T,sep="\t")$V3
)
}
has_qq## [1] TRUE TRUE TRUE FALSE FALSENow we can take off the [1:5] limiter, and do the whole set:
# listing files
filez <- list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")
# initializing vectors
has_qq <- vector("logical",length(filez))
# looping through files
for (i in seq_along(filez) ) {
# calculating length and storing it
has_qq[i] <- grepl( "QQ", read.delim(filez[i],
header=F,as.is=T,sep="\t")$V3
)
}
has_qq[1:10]## [1] TRUE TRUE TRUE FALSE FALSE TRUE FALSE FALSE TRUE FALSEHow many proteins have “QQ”? Cool trick for logical vectors, sum.
## [1] 148## has_qq
## FALSE TRUE
## 95 148Can also ggplot it, for fun:
library(ggplot2)
ggplot( data.frame(qq_status=has_qq) )+
theme_classic()+
aes(x=qq_status)+
geom_bar()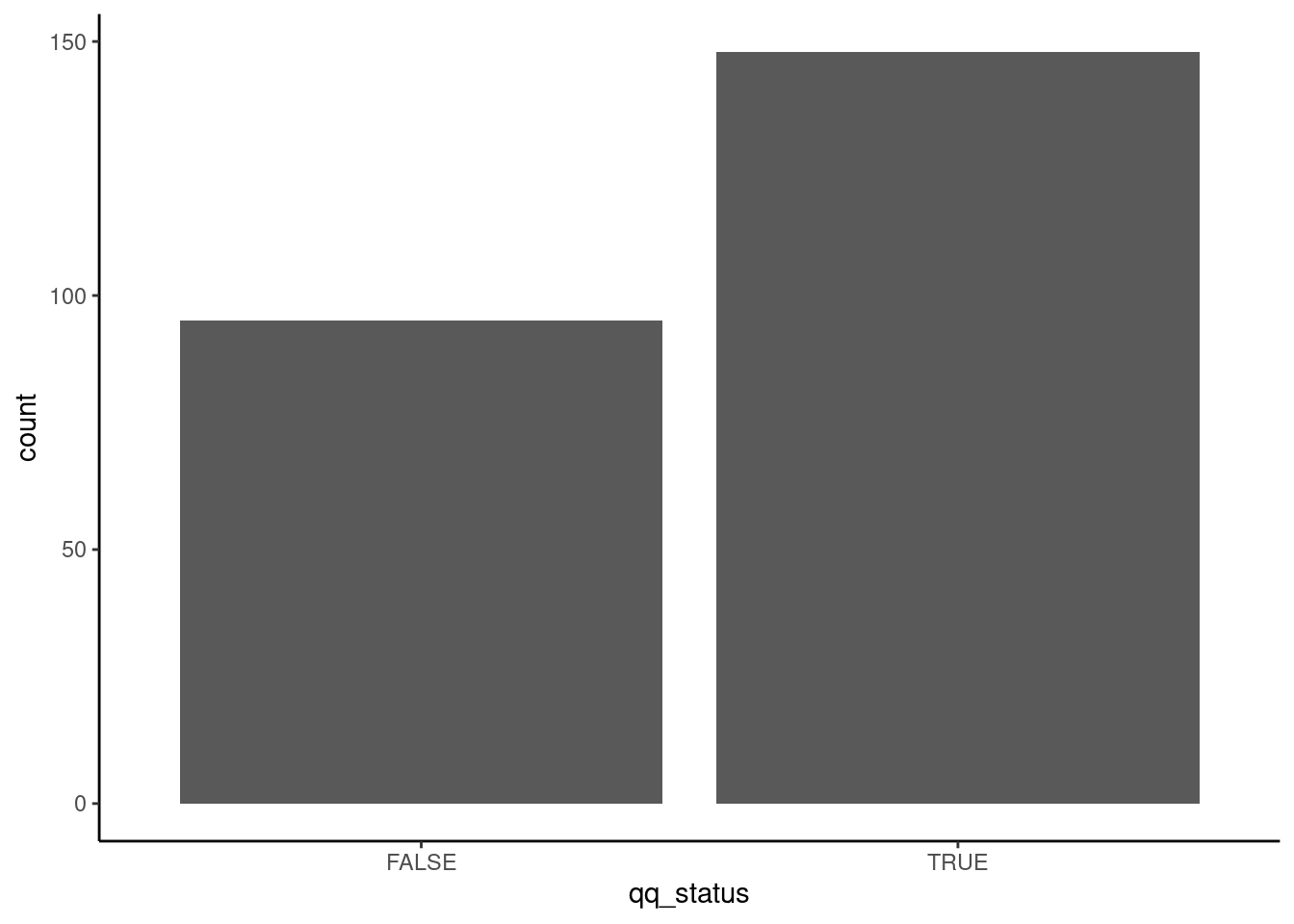
4.2.5 Extending the workflow
How do you think about/plan the workflow we’ve built?
How is it organized?
One way is to flatten out all the tasks, and to script each individual task every time it is done. This requires the author, user, and reader to understand a lot of complexity.
Another way to approach this is to cluster them into a hierarchy of modules.
In this organization, the analyst can operate at levels of steps, modules, and specific instructions, depending on what is needed.
Organizing your workflows into composable modules lets you extend these to un-ancipiated complexity. You could imaging using these steps or modules:
in new ways by composing the elements together, to analyze a different source of proteins, with a new analysis, with similar plots:
Where maybe these are how the inputs/outputs are defined:
reading_proteins:
input: directory path of proteins to read
output: protein sequences
calculating lengths:
input: protein sequences
output: numeric vector, of lengths
finding a motif:
input: protein sequences
output: numeric vector, of presence/absence4.2.6 Conclusion
Write this up! Edit your rmarkdown file to use
4.2.6.1 headers
italics
bold statements
, reference previous figures, or make additional plots, or list out filenames to support your claim. Maybe just spend five minutes for now, but get used to writing your notes down so you save them.
Ideas:
- You can analyze strings using functions like nchar or grepl.
- You can use loops to organize repetitive tasks, such as working through a
large list of files and reading them.
- Modular design of workflows, using functions or just organized and
commented code, can help you extend your analysis to handle new
questions as they arise
4.2.7 Totally optional supplements, for your edification
4.2.7.1 stringr and the tidyverse
The tidyverse includes a package stringr. This cleans up some of these
basic string operations, makes them more standardized, and gives them nice
standardized names. It’s worth using, but you should know about the other
base R functions!
For example the above,
datar <- data.frame(stringsAsFactors=F,
filenames=c(
"210607_pilot_transformation_works.jpeg",
"210609_transformation_seems_to_work.jpeg",
"210610_failed_gel_images.jpeg"
)
)
datar$dates <- str_extract(string=datar$filenames,pattern="\\d\\d\\d\\d\\d\\d_")
datar## filenames dates
## 1 210607_pilot_transformation_works.jpeg 210607_
## 2 210609_transformation_seems_to_work.jpeg 210609_
## 3 210610_failed_gel_images.jpeg 210610_There are many options, type str_ and then TAB.
For much much more about strings, check out Wickham’s R4DS book. Especially for the special characters part
Or even cleaner using tidyverse verbs:
## ── Attaching packages ───────────────────────────────────────────────────────────────────────────────────── tidyverse 1.3.0 ──## ✔ tibble 3.0.4 ✔ purrr 0.3.4
## ✔ tidyr 1.1.2 ✔ dplyr 1.0.2
## ✔ readr 1.4.0 ✔ forcats 0.5.0## ── Conflicts ──────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()tibble(filenames=c(
"210607_pilot_transformation_works.jpeg",
"210609_transformation_seems_to_work.jpeg",
"210610_failed_gel_images.jpeg"
)
) %>%
separate(col=filenames,sep="_",into=c("date","rest"),extra="merge") %>%
separate(col=rest,sep="\\.",into=c("name","type")) ## # A tibble: 3 x 3
## date name type
## <chr> <chr> <chr>
## 1 210607 pilot_transformation_works jpeg
## 2 210609 transformation_seems_to_work jpeg
## 3 210610 failed_gel_images jpegI’d recommend you use the tidyverse-style manipulations - they’re powerful.
But you should be familiar with base R, to fix bugs.
4.2.7.2 More tidyverse demo
tidyverse is pretty neat at doing these things in simpler, more readable
code. Here’s how you might approach the motif-finding task in that paradigm:
1st - list files, but set it in a tibble:
tibble(
filenames=list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")
)## # A tibble: 243 x 1
## filenames
## <chr>
## 1 data/viral_structural_proteins/viral_proteins_000.tsv
## 2 data/viral_structural_proteins/viral_proteins_001.tsv
## 3 data/viral_structural_proteins/viral_proteins_002.tsv
## 4 data/viral_structural_proteins/viral_proteins_003.tsv
## 5 data/viral_structural_proteins/viral_proteins_004.tsv
## 6 data/viral_structural_proteins/viral_proteins_005.tsv
## 7 data/viral_structural_proteins/viral_proteins_006.tsv
## 8 data/viral_structural_proteins/viral_proteins_007.tsv
## 9 data/viral_structural_proteins/viral_proteins_008.tsv
## 10 data/viral_structural_proteins/viral_proteins_009.tsv
## # … with 233 more rows2nd - pipe that into a mutate call, where you use map on filenames.
map is like lapply. It takes whatever you have on the left, and puts
it one by one into the function on the right, along with optional arguments
that you list after it. Here, we set col_names to false,
analogous to header in read.delim.
tibble(
filenames=list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")
) %>%
mutate(rawfiles=map(filenames,read_tsv,col_names=F)) ## # A tibble: 243 x 2
## filenames rawfiles
## <chr> <list>
## 1 data/viral_structural_proteins/viral_proteins_000.tsv <tibble [1 × 3]>
## 2 data/viral_structural_proteins/viral_proteins_001.tsv <tibble [1 × 3]>
## 3 data/viral_structural_proteins/viral_proteins_002.tsv <tibble [1 × 3]>
## 4 data/viral_structural_proteins/viral_proteins_003.tsv <tibble [1 × 3]>
## 5 data/viral_structural_proteins/viral_proteins_004.tsv <tibble [1 × 3]>
## 6 data/viral_structural_proteins/viral_proteins_005.tsv <tibble [1 × 3]>
## 7 data/viral_structural_proteins/viral_proteins_006.tsv <tibble [1 × 3]>
## 8 data/viral_structural_proteins/viral_proteins_007.tsv <tibble [1 × 3]>
## 9 data/viral_structural_proteins/viral_proteins_008.tsv <tibble [1 × 3]>
## 10 data/viral_structural_proteins/viral_proteins_009.tsv <tibble [1 × 3]>
## # … with 233 more rows3rd - we then unnest the rawfiles. This takes a column of tibbles and makes
it magically just line up with the top level tibble.
Now we’ll also save it as variable datar.
datar <- tibble(
filenames=list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")
) %>%
mutate(rawfiles=map(filenames,read_tsv,col_names=F)) %>%
unnest(rawfiles)Then we can process on datar. Note the use of mutate, map, and
wrapping map in unlist to just return the lengths instead of a list of
lengths.
datar %>%
mutate(
protein_length=unlist(map(X3,nchar)),
qq_present=unlist(map(X3,grepl,pattern="QQ")),
)## # A tibble: 243 x 6
## filenames X1 X2 X3 protein_length qq_present
## <chr> <chr> <chr> <chr> <int> <lgl>
## 1 data/viral_str… Q8JUX5.3 … Chikungun… MEFIPTQTFYNR… 1248 TRUE
## 2 data/viral_str… YP_009507… Everglade… MFPFQPMYPMQP… 1254 TRUE
## 3 data/viral_str… NP_042704… African s… MFNIKMTISTLL… 177 TRUE
## 4 data/viral_str… NP_042736… African s… MEAVLTKLDQEE… 137 FALSE
## 5 data/viral_str… P27413.1 … Rabbit he… MAFLMSEFIGLG… 117 FALSE
## 6 data/viral_str… NP_042800… African s… MDTETSPLLSHN… 117 TRUE
## 7 data/viral_str… NP_050192… Human bet… MDLKAQSIPFAW… 858 FALSE
## 8 data/viral_str… Q306W5.1 … Eastern e… MFPYPTLNYSPM… 1242 FALSE
## 9 data/viral_str… NP_040824… Venezuela… MFPFQPMYPMQP… 1255 TRUE
## 10 data/viral_str… YP_003987… Acanthamo… MNYSLEDLPNSG… 234 FALSE
## # … with 233 more rowsAnd then we can pipe it into a ggplot:
datar %>%
mutate(
protein_length=unlist(map(X3,nchar)),
qq_present=unlist(map(X3,grepl,pattern="QQ")),
) %>%
ggplot()+
aes(x=qq_present)+
geom_bar()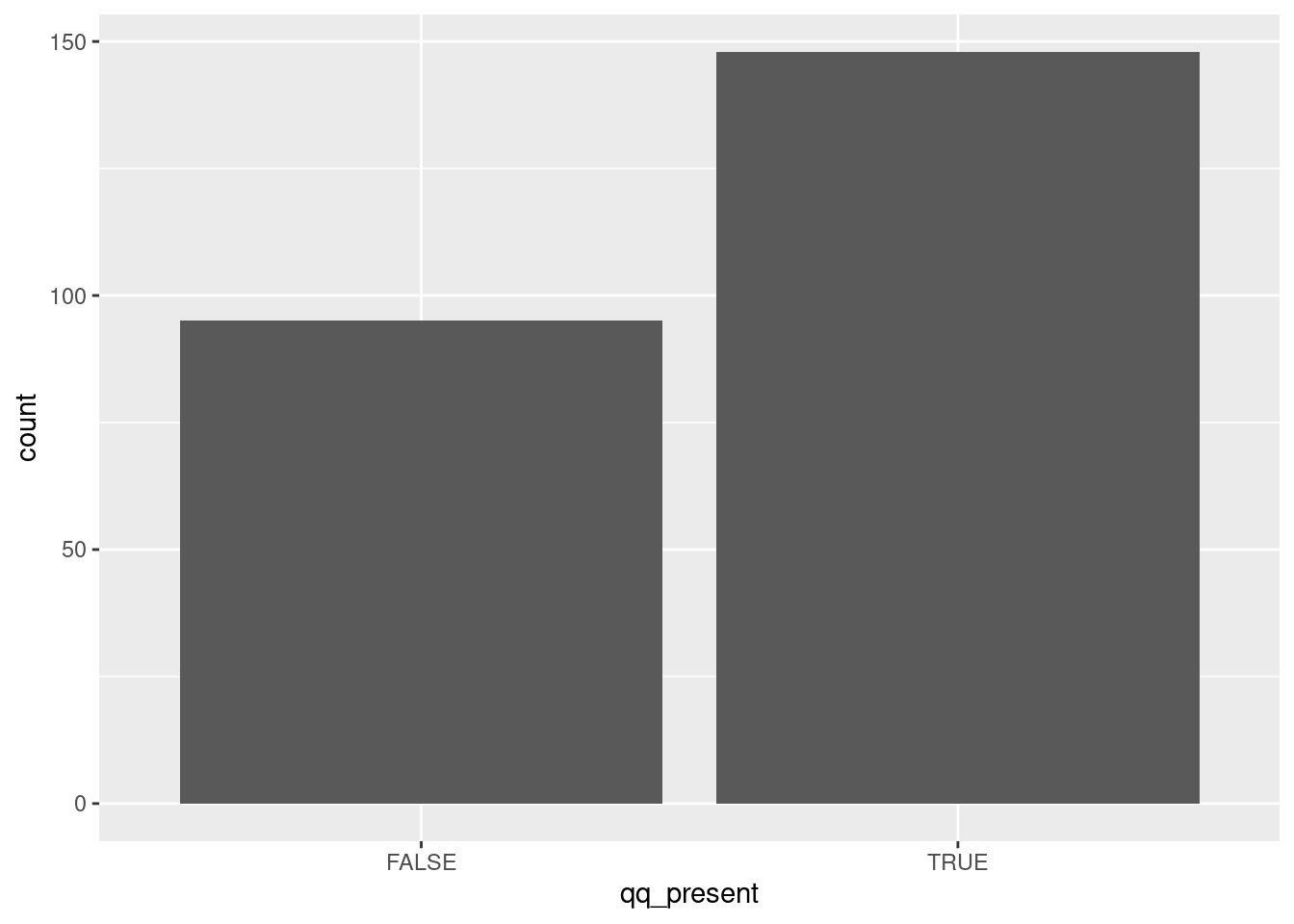
And the entire code:
tibble(
filenames=list.files(path="data/viral_structural_proteins",
full.names=T, pattern=".*tsv")
) %>%
mutate(rawfiles=map(filenames,read_tsv,col_names=F)) %>%
unnest(rawfiles) %>%
mutate(
protein_length=unlist(map(X3,nchar)),
qq_present=unlist(map(X3,grepl,pattern="QQ")),
) %>%
ggplot()+
aes(x=qq_present)+
geom_bar()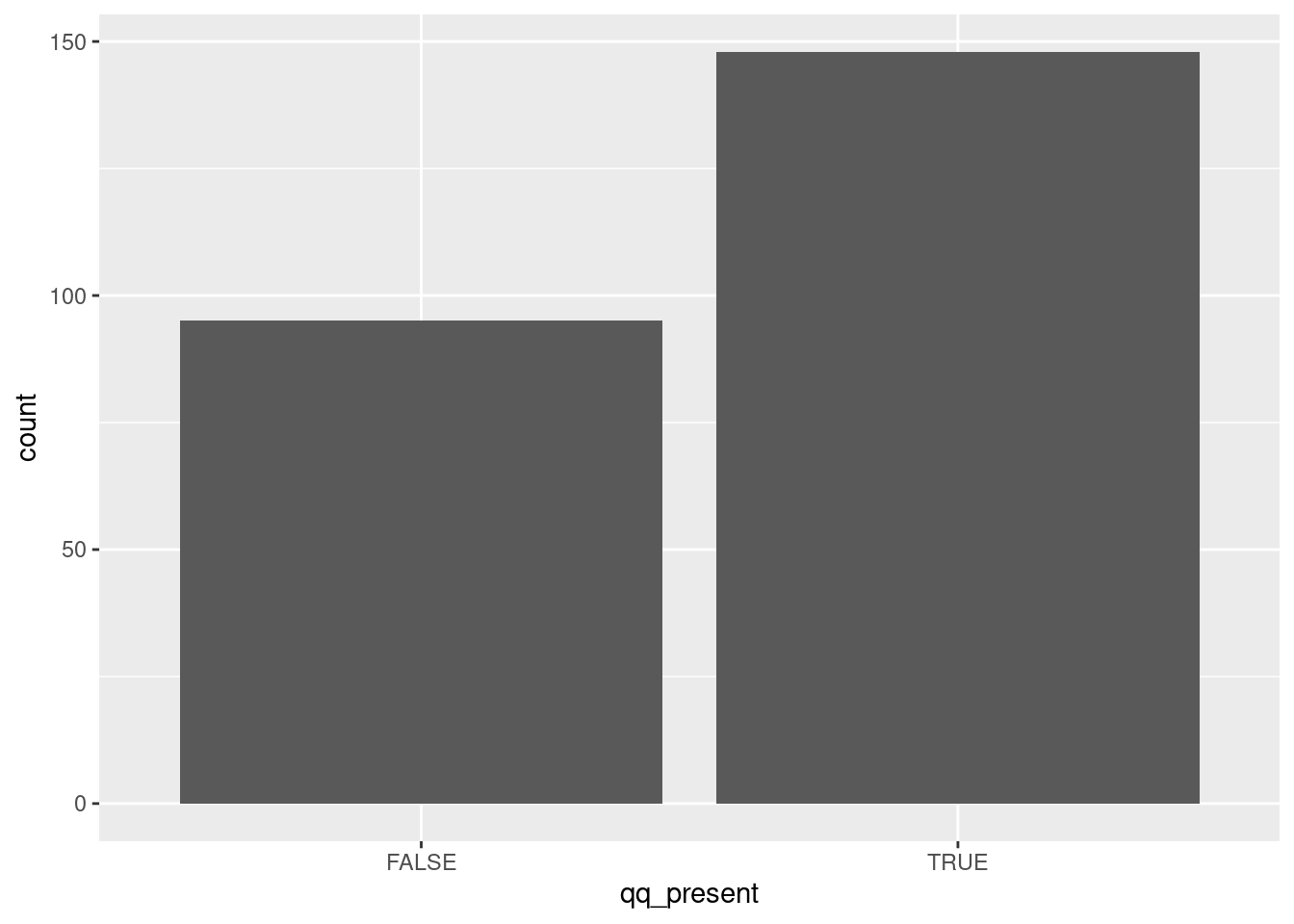
I think that’s tidy.
One crucial aspect missing is the actual bash commands used. I did not save them as a script, and I should go back, save them in a script, and re-run that to make sure I get the same result!↩
The formats you’re likely going to have to deal with are CSV, TSV, FASTQ, FASTA, SAM - at least. ↩
…since we’re just demonstrating R coding and not representing ourselves as being familiar enough with the field to properly interpret these results. I have no idea what occurance of this would mean or not mean, or are we doing any sort of statistics or comparisons to null expectations.↩
Licensed Creative Commons Attribution-NonCommercial-ShareAlike 4.0